尔云间 一个专门做科研的团队

scATAC-seq是什么?
可以用来分析什么?
与scRNA-seq的区别是什么呢?
之前小云分享过单细胞测序(scRNA-seq)、bulk RNA测序和空间转录组测序(stRNA-seq),三者联合分析的生信思路。顺便回顾一下这三者之间区别:
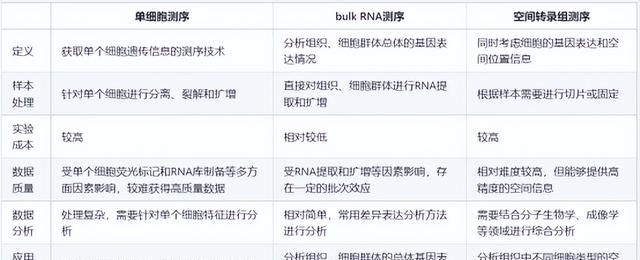
今天小云又发现一篇4种组学的生信思路,除了上面提到的三种,还有scATAC-seq。
(1)scATAC-seq是什么?
scATAC-seq是单细胞染色质可及性测序技术(Single-cell Assay for Transposase-Accessible Chromatin using sequencing)的缩写。它可以用于评估单个细胞的基因组范围内染色质开放性和转录因子结合,从而确定单个细胞基因表达和调控的情况。
具体来说,scATAC-seq利用一种名为转座酶的酶将DNA片段插入到基因组中的开放染色质区域,随后进行PCR扩增并进行高通量测序,以获取每个单个细胞的染色质可及性图谱。这种技术主要用于研究不同细胞类型之间的差异、不同时期细胞内的染色质重构、结构变异等方面的问题。
(2)scATAC-seq与普通的染色质可及性测序的区别?
相比于传统的大规模染色质可及性测序技术,scATAC-seq具有分辨率高、灵敏度高、样本数量需求低等优点。同时,由于该技术可以对单个细胞进行测序,并生成单个细胞特定的染色质图谱,因此可以避免批次效应和混杂效应的问题。这使得它成为研究单个细胞的转录因子调控和染色质开放性的理想工具。
(3)scATAC-seq与scRNA-seq、空间转录组测序(stRNA-seq)的区别?
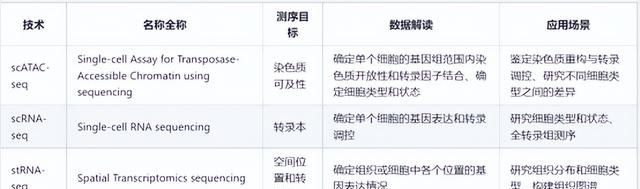
可以看到,这三种技术各有特点,适用于不同的研究问题。scATAC-seq主要用于研究染色质重构与转录调控,而scRNA-seq则主要用于研究基因表达和转录调控,stRNA-seq主要用于研究组织中的基因表达情况。同时,scATAC-seq可以确定单个细胞的基因组范围内染色质开放性和转录因子结合,而scRNA-seq和stRNA-seq则只能确定单个细胞的转录本或空间位置上的基因表达信息。
如果手里有样本+经费,可以直接做测序发文。当然随着测序数据越来越多,直接对公共数据库中现成的数据进行分析,也是一种迂回策略~(不知道如何找新思路或创新升级的可以找小云,各种个性化分析思路等你来挑~)
今天小云给大家分享一篇合了bulk RNA-seq、scRNA-seq、scATAC-seq 、stRNA-seq四种组学分析为一文的生信文章,除了组织数据还有外泌体数据,还用到了6种细胞死亡类型相关基因,多组学分析直接拿下8分+纯生信文章!一起来学习一下吧~

文章题目:程序性细胞死亡分析:利用血源性外泌体转录组学信息和肿瘤微环境中的单细胞多组学数据构建肾透明细胞癌(KIRC)诊断模型
影响因子:8分+
发表时间:2023年4月
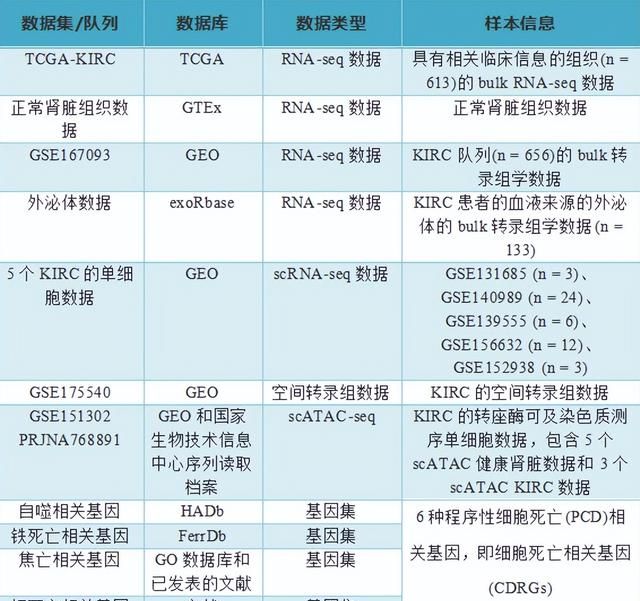
数据信息
研究思路
收集细胞凋亡、坏死性凋亡、自噬、焦亡、铁死亡、铜死亡6类细胞死亡相关基因(CDRGs)。下载来自exoRBase数据库的血源性外泌体RNA测序(RNA-seq)数据、来自TCGA和GTEx数据库的组织RNA-seq数据以及来自GEO数据库的单细胞RNA测序(scRNA-seq)数据。将来自exoRBase和TCGA数据库的KIRC队列的差异表达基因(DEGs)与来自单细胞数据集的CDRGs和DEGs相交,利用临床指标和机器学习方法进一步筛选候选生物标志物基因,从而构建KIRC的诊断模型。最后,利用scRNA-seq、单细胞转座酶可及染色质测序(scATAC-seq)和空间转录组学测序(stRNA-seq)数据,研究了关键基因的潜在机制及其在肿瘤微环境中的作用。

图1. 分析流程图
主要研究结果
从GEO数据库中下载5个单细胞数据集进行分析,用Seurat包鉴定出54个细胞簇和10种主要细胞类型,包括肿瘤上皮细胞、正常上皮细胞、内皮细胞(Endo)、成纤维细胞(Fib)、T细胞、B细胞、巨噬细胞(Mac)、单核细胞(Mono)、自然杀伤细胞(NK)和嗜碱性细胞(Baso)(图2A)。各细胞群的特异性标记和相对丰度如图2B所示。使用UMAP可视化了每种主要细胞类型及其起源的分布(图2C)。随后,分析scRNA-seq数据集中每个细胞簇的癌症样本和对照样本之间差异表达基因(DEGs),肿瘤和正常上皮细胞之间的DEGs数量最高(图2D)。
接下来,分析TCGA队列与GTEx中健康样本的转录组数据,并使用exoRBase数据库分析了健康对照组和KIRC患者的人血源性外泌体的RNA-seq数据,并展示DEGs(图2E),对DEGs进行KEGG功能富集分析 (图2F)。
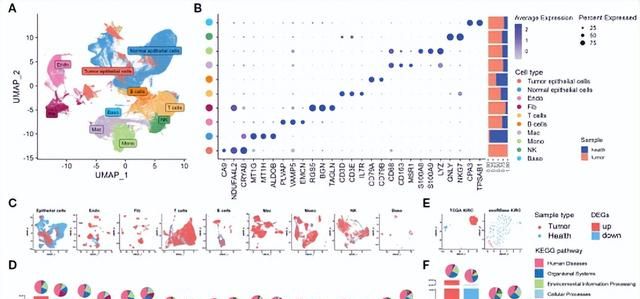
图2. KIRC单细胞RNA测序(scRNA-seq)和bulk RNA测序(RNA-seq)图谱
2. 6种细胞死亡相关基因(CDRGs)在KIRC中的表达模式
纳入凋亡、坏死、自噬、焦亡、铁死亡、铜死亡等6种PCDs及其相关基因CDRGs进行分析 (图3A)。在单细胞和RNA-seq数据集中,大多数CDRGs是上调的DEGs(图3B)。说明CDRGs的表达水平不同程度地增强。
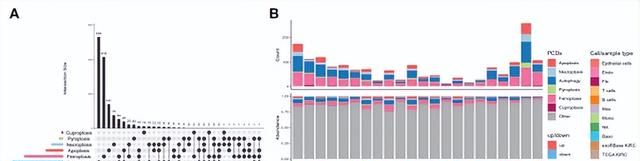
图3. KIRC中程序性细胞死亡的分布特征
接下来,作者将exoRBase KIRC中上调(图4A)和下调(图4B)的DEGs与从单细胞和TCGA数据集中获得的CDRGs和DEGs取交集,得到了53个候选生物标记基因。其中有32个基因与患者的临床分期或生存结果密切相关(图4D)。32个基因中有13个基因同时与KIRC的临床分期和生存结果相关。
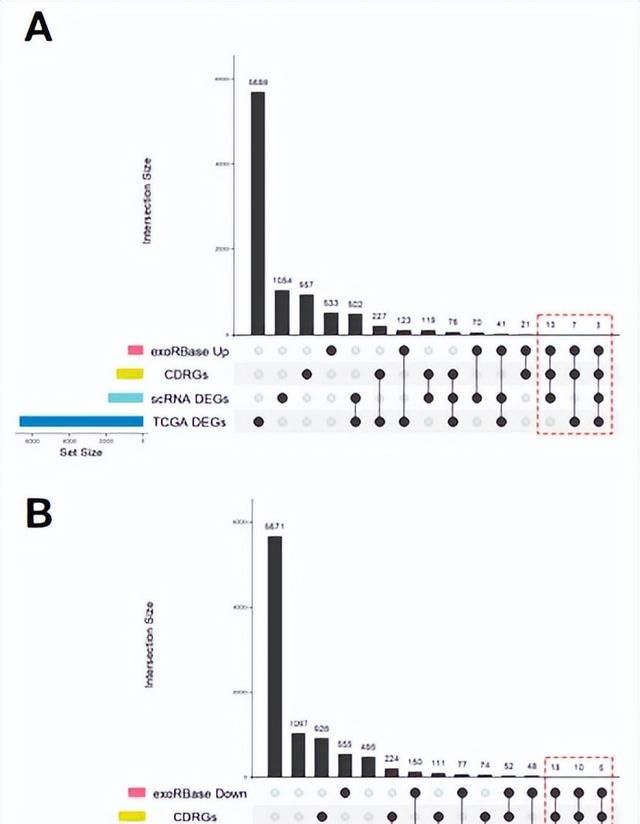
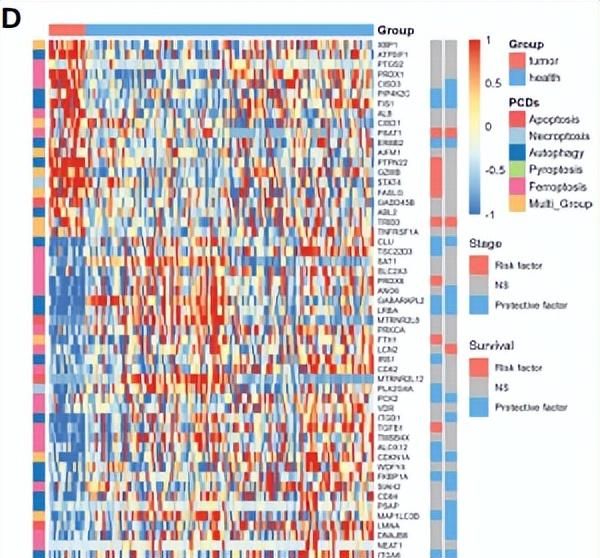
图4. 候选生物标记基因的筛选
3. 基于13个基因构建诊断模型并验证
使用13个关键基因构建KIRC的诊断模型,发现诊断模型exoRbase数据(图5A)和TCGA KIRC队列中显示出良好的诊断结果(图5B)。
然后,作者分析了scATAC数据集中的63,489个KIRC病例细胞,根据代表性标记基因的平均启动子活性确定了15种主要细胞类型(图5C, D),发现TRIB3在肿瘤上皮细胞中的表达高于正常上皮细胞。同时,其染色质可及性显著增加(图5E)。
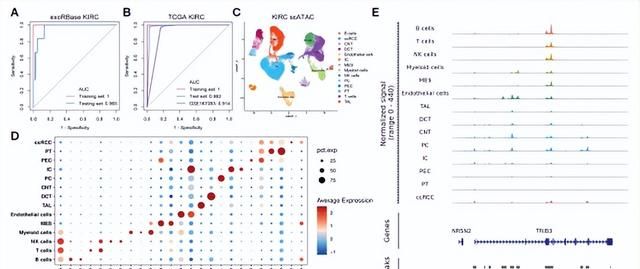
图5.基于13个基因的诊断模型和单细胞染色质可及性测序(scATAC-seq)分析
4. 对TRIB3high肿瘤上皮细胞的分析
对TRIB3的分析表明,该基因与KIRC的TNM分期呈正相关(图6A-C)。scRNA数据集分析发现,TRIB3high亚群出现在肿瘤上皮细胞中(图6D),与不同细胞类型之间具有相关性 (图6E)。伪时间分析表明,这种细胞亚群可能是肿瘤干细胞(图6F)。细胞间通讯分析表明,TRIB3高表达的肿瘤细胞与Mac和T细胞的存在相互作用(图6G)。
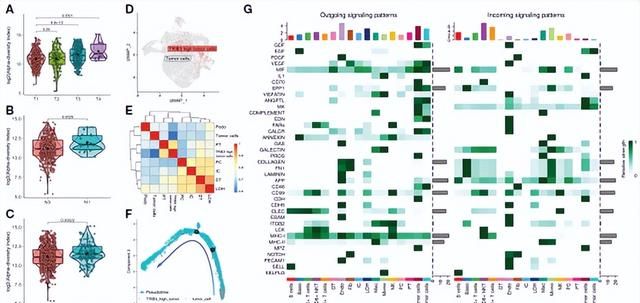
图6. KIRC患者TRIB3表达的临床特征及TRIB3高表达肿瘤细胞亚群的特征
与其他细胞簇相比,TRIB3high肿瘤上皮细胞主要富集于凋亡、铁死亡、核糖体和溶酶体信号通路(图7A、B)。最后,空间转录组学分析证实TRIB3high亚群在肿瘤组织中高度富集(图7C)。
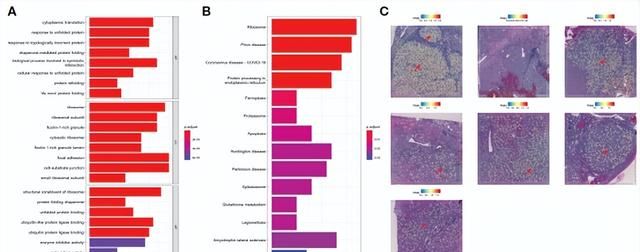
图7. TRIB3high肿瘤细胞的功能分析及空间定位
文章小结
这篇文章的亮点就在于用到了单细胞测序、bulk RNA测序、空间转录组测序、scATAC-seq四种组学数据进行分析,除了组织数据还有外泌体数据,还用到了6种细胞死亡类型相关基因,其实分析内容并没有很多很复杂。
上述4种王炸组合叠加,多组学分析+多种细胞死亡,即使没有加实验验证,也能够发到8分+,如此创新和性价比高的分析思路,赶快学起来吧!
如果你还苦恼于生信分析没有思路,或者嫌分析方法太过简单、太过老套,想要创新思路的,或者对多组学联合分析/多种细胞死亡类型感兴趣的小伙伴快来联系小云吧!

- 单细胞+bulk RNA-seq+空间转录组
- 转录组+GWAS联合分析
- 转录+蛋白+表观多组学分析+免疫分析
- 临床试验+多组学联合生信分析筛选标志物
- 生物标志物筛选+GWAS等多组学分析+预后分析
- 多组学分析+预后模型构建+简单验证